la présence et la densité des ondes F du répéteur sont principalement liées au degré de perte de LMN et elles ne montrent aucune corrélation évidente avec le dysfonctionnement du système UMN
En neuroscience, une onde F est le deuxième des deux changements de tension observés après l'application d'une stimulation électrique à la surface de la peau au-dessus de la région distale d'un nerf.
Les ondes F sont souvent utilisées pour mesurer la vitesse de conduction nerveuse et sont particulièrement utiles pour évaluer les problèmes de conduction dans la région proximale des nerfs (c'est-à-dire des portions de nerfs proches de la moelle épinière).
Elles sont quasi universellement utilisées pour le diagnostic de la SLA. En même temps les patients de la SLA se sont souvent plaint de la qualité de cet examen, qui donne régulièrement lieu à des diagnostics incorrects. Un nouvel article par Akarsu et ses collègues montre qu'effectivement d'autres techniques, utilisant le même outillage sont plus précises. Il se pourrait même qu'à terme on réexamine l'affirmation que la SLA est une maladie affectant à la fois les motoneurones supérieurs et inférieurs.
Dans une étude d'onde F typique, un fort stimulus électrique est appliqué sur la surface de la peau au-dessus de la partie distale d'un nerf, de sorte que l'impulsion se déplace à la fois distale (vers la fibre musculaire) et proximale (jusqu'aux neurones moteurs de la moelle épinière).
Ces impulsion sont également appelées orthodromique et antidromique, respectivement. Lorsque le stimulus orthodromique atteint la fibre musculaire, il provoque une forte réponse en M indiquant une contraction musculaire.
Lorsque le stimulus antidromique atteint les corps cellulaires du motoneurone, une petite partie de celui-ci se retourne contre elle et une onde orthodromique redescend du nerf vers le muscle.
Ce stimulus réfléchi évoque une faible proportion des fibres musculaires, ce qui entraîne un second groupe de potentiels d’action presque simultanés provenant de plusieurs fibres musculaires situées dans la même zone, appelé onde F.
Des biomarqueurs électrophysiologiques ont permis de réaliser d’énormes travaux de détection et de quantification du dysfonctionnement du neurone moteur supérieur (UMN) et du neurone moteur inférieur (LMN) dans la sclérose latérale amyotrophique.
L'index neurophysiologique et les méthodes d'estimation du nombre d'unités motrices (MUNE) ont été largement utilisés en tant que biomarqueurs potentiels de la perte de LMN.
L'indice neurophysiologique a été suggéré pour démontrer la perte de LMN chez les patients atteints de SLA, même dans les muscles présymptomatiques, et s'est révélé sensible à la détection de la progression de la maladie.
Bien que plusieurs méthodes MUNE et une stimulation magnétique transcrânienne à impulsions uniques et à impulsions appariées aient été proposées depuis l'invention de la première technique en 1971, aucune d'entre elles n'a été acceptée comme méthode standard en raison des diverses limitations inhérentes à la technique.
Dans la SLA, maladie affectant à la fois les UMN et les LMN, des mécanismes corticaux et périphériques ont été proposés pour expliquer les anomalies de l’onde F.
Une augmentation du nombre d'ondes F de répéteur en présence d'une implication clinique de l'UMN a été rapportée dans la SLA.
En revanche, il a été constaté que les muscles atrophiés, plus marqués dans la région thénar, généraient davantage d’ondes F de répéteur, ce qui concorde avec le phénomène de division de la main qui se produit dans la même maladie.
Globalement, le mécanisme de génération des ondes de répéteur est toujours discuté.
Dans la présente étude, les auteurs visaient à étudier les ondes F répétées dans les muscles thénar et hypothénar des patients atteints de SLA et leur corrélation avec d’autres marqueurs électrophysiologiques afin de mieux comprendre la dominance du dysfonctionnement de l’UMN ou du LMN dans le mécanisme de leur émergence.
Leurs résultats, dans leur ensemble, suggèrent que la présence et la densité des ondes F du répéteur sont principalement liées au degré de perte de neurones moteurs inférieurs.
En réponse à la perte progressive de motoneurones, la réinnervation intervient pour compenser et les résultats de ces processus doubles établissent les caractéristiques diagnostiques de la SLA.
Le nombre réduit de motoneurones dans la génération des ondes F donne lieu à un plus grand nombre d'ondes F de répéteur.
D'autre part, les grandes ondes F et les ondes F géantes du répéteur ont été associées à des unités motrices ré-innervées.
Une étude antérieure avait montré que la fréquence des ondes F de répéteur était augmentée chez les patients atteints de SLA présentant des signes pyramidaux par rapport au groupe non pyramidal. Les auteurs ont donc divisé les groupes de patients en fonction de la présence ou de l’absence de signes pyramidaux et n’ont pas utilisé d’outil quantitatif pour déterminer l’atteinte du tractus cortico-spinal.
Les études sur les ondes F dans des maladies impliquant uniquement des UMN, telles que la sclérose en plaques et les maladies cérébrovasculaires, ont montré une augmentation de la persistance, de l’amplitude, de la durée et de la latence de l’onde F, mais aucune de ces études n’a étudié les ondes F répétées.
Selon leurs résultats, les scores de somme ALSFRS-R et MRC n’étaient pas corrélés avec les paramètres de répétition des ondes F.
Ces scores cliniques fournissent une évaluation fonctionnelle globale chez les patients atteints de SLA.
De plus, le score ALSFRS-R UL, le sous-score de ALSFRS-R adressant la fonction des membres supérieurs, n'a également révélé aucune corrélation.
Ceci suggère que les scores cliniques reflètent moins la perte motoneuronale, probablement en raison de la capacité de compensation rémanente du système moteur, et que les ondes F de répéteur pourraient fournir une mesure antérieure de la dégénérescence des motoneurones, comme la plupart des méthodes électrophysiologiques consacrées à ce sujet.
Conclusion
Leurs résultats, dans leur ensemble, suggèrent que la présence et la densité des ondes F du répéteur sont principalement liées au degré de perte de LMN et elles ne montrent aucune corrélation évidente avec le dysfonctionnement du réseau UMN.
Publicité

Ce livre retrace les principales réalisations de la recherche sur la SLA au cours des 30 dernières années. Il présente les médicaments en cours d’essai clinique ainsi que les recherches en cours sur les futurs traitements susceptibles d’ici quelques années, d’arrêter la maladie et de fournir un traitement complet en une décennie ou deux.
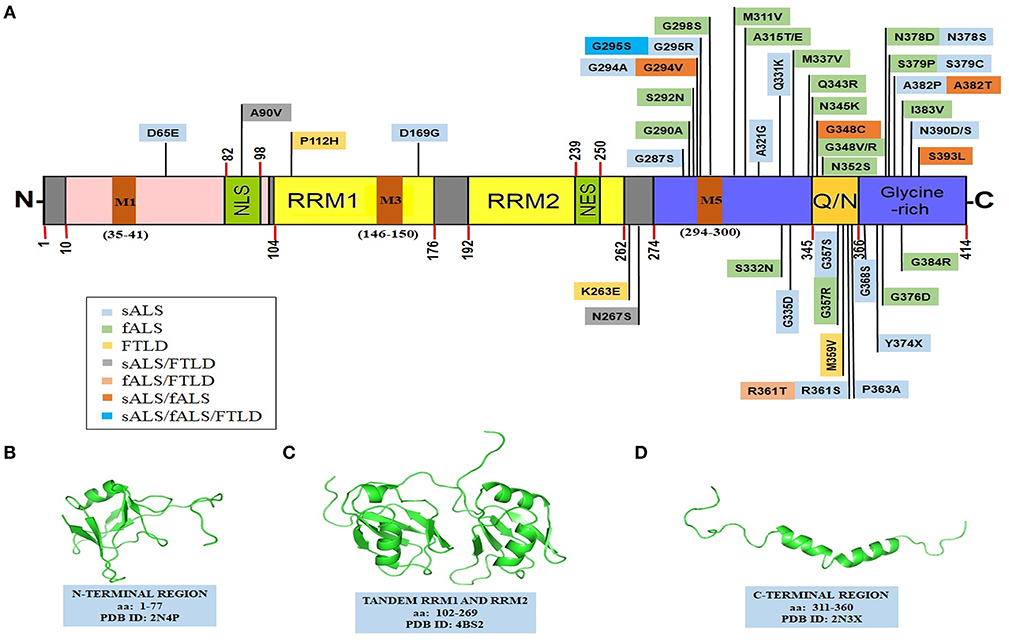 Les auteurs ont montré qu'en présence d’une acidification, même très faible, le TDP-43tRRM se replie complètement et s'oligomérise pour former une «forme β» riche en feuillets β. La forme β a une structure ordonnée et stable qui ressemble aux fibrilles amyloïdes que l'on trouve dans la maladie d'Alzheimer!
Les auteurs ont montré qu'en présence d’une acidification, même très faible, le TDP-43tRRM se replie complètement et s'oligomérise pour former une «forme β» riche en feuillets β. La forme β a une structure ordonnée et stable qui ressemble aux fibrilles amyloïdes que l'on trouve dans la maladie d'Alzheimer!
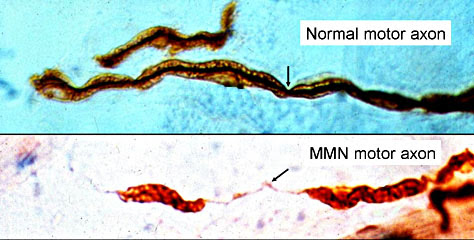 La nature exacte des problèmes apparaît cependant être variable dans ces différentes maladies; par exemple les concentrations de ganglioside
sont réduites dans la maladie de Parkinson et la maladie de Huntington, mais augmentées dans la maladie d’Alzheimer et y a des altérations dans les deux directions pour la SLA.
La nature exacte des problèmes apparaît cependant être variable dans ces différentes maladies; par exemple les concentrations de ganglioside
sont réduites dans la maladie de Parkinson et la maladie de Huntington, mais augmentées dans la maladie d’Alzheimer et y a des altérations dans les deux directions pour la SLA. Par BrainsRusDC - Travail personnel, CC BY 4.0, https://commons.wikimedia.org/w/index.php?curid=64271015
Par BrainsRusDC - Travail personnel, CC BY 4.0, https://commons.wikimedia.org/w/index.php?curid=64271015 Source: Blausen.com staff (2014). "Medical gallery
Source: Blausen.com staff (2014). "Medical gallery

