Abnormal protein aggregation within cells is a recurring phenomenon in Parkinson's disease (PD), Alzheimer's disease (AD), and amyotrophic lateral sclerosis (ALS). Current approaches use antibodies to target these aggregates, but this is a rudimentary approach, as little is known about the causes of their formation, or whether they are the cause or consequence of the disease.
Cells are an incredibly crowded environment, and their molecules undergo Brownian motion, which thwarts their biological function. Making the cell less dense and more soluble would certainly alleviate some molecular problems. There are various approaches, including those that use phase transitions.
Recent research sheds surprising light on the dynamic relationship between mitochondrial activity, ATP levels, and neuronal cytoplasmic fluidity, all of which play a critical role in controlling protein aggregation.
The researchers used mouse giant goblet cell cultures to analyze presynaptic viscosity using real-time confocal microscopy. These cells are characterized by large glutamatergic nerve terminals, ideally suited for real-time imaging. Rather than focusing on individual proteins, the team took a holistic approach, using a technique called fluorescence recovery after photobleaching (FRAP) of soluble green fluorescent protein (cGFP) to assess the overall viscosity of the axonal cytosol.
Cytosolic viscosity can reflect the extent of protein aggregation; Greater aggregation means less free diffusion of cGFP, indicating a more "solidified" cytosol.
Synapses are hotspots for mitochondria, which provide the ATP needed for neurotransmission. By labeling active mitochondria and comparing their location to cGFP mobility, the study revealed that regions with greater mitochondrial activity exhibited higher cytosolic fluidity. This suggests a direct link between ATP production and the maintenance of a more soluble and functional presynaptic environment.
To further investigate this, the team inhibited mitochondrial function using FCCP and other mitochondrial blockers. As ATP production decreased, cGFP diffusion decreased sharply, suggesting that the cytosol was becoming more viscous due to protein aggregation. It is important to note that this effect was specific to mitochondrial inhibition: blocking glycolysis had little effect.
Even components of the synaptic release mechanism, such as synaptic vesicles (SVs) and active zones (AZs), exhibited reduced mobility under mitochondrial stress, reinforcing the idea that energy depletion disrupts the fluid phase of the cytoplasm.
To test whether ATP could restore the altered cytosol state, the researchers administered ATP directly to neurons. They found that ATP not only restored cGFP diffusion but also reduced the size and number of protein aggregates. To test whether enhancing endogenous ATP production could mitigate the protein aggregation linked to mitochondrial dysfunction, the researchers turned to NMN, a molecule known to boost NAD⁺ levels and support mitochondrial health.
They treated neurons with NMN and observed the following key outcomes:
Partial restoration of cytoplasmic fluidity: In neurons with compromised mitochondrial activity (such as those derived from PARK2 or TDP-43 mutant patients), NMN treatment significantly improved the diffusion of soluble proteins like cGFP. While not as dramatic as direct ATP infusion, NMN nonetheless reduced cytosolic viscosity.
Reduction in aggregate burden: In both mouse neurons under mitochondrial stress and hiPSC-derived human neurons from neurodegenerative disease patients, NMN treatment lowered the accumulation of insoluble protein aggregates.
Improved ATP levels: NMN supplementation helped increase intracellular ATP concentrations, presumably by enhancing mitochondrial NAD⁺-dependent enzymatic activity, which supports oxidative phosphorylation.
These results suggest that NMN supports the same protective pathway as ATP, but indirectly, by restoring mitochondrial capacity to generate ATP and maintain a more fluid intracellular environment.
The mechanism appears to be biophysical rather than biochemical: ATP acts as a hydrotrope, a molecule that keeps other proteins dissolved and prevents them from forming aggregates.
The researchers then examined whether this principle held true for specific proteins involved in neurodegenerative diseases, including:
α-synuclein (mutant SNCA and SNCA-A53T), PARK2 – Parkinson's disease
APP, Amyloid, Tau – Alzheimer's disease
TDP-43 – ALS
These purified proteins were able to undergo liquid-liquid phase separation (LPS) and form condensates in vitro. ATP was able to dissolve many of these condensates in a concentration-dependent manner, although mutant or misfolded versions (e.g., SNCA-A53T) required higher ATP concentrations to dissolve.
When the aggregates were left to incubate for longer, some (notably SNCA-A53T) began to form protofibrils, elongated, fibril-like structures similar to those observed in real-life pathology. Here again, ATP could reverse this phenomenon, but with reduced efficiency.
Even under crowded conditions (mirrored by the addition of PEG), ATP retained some ability to prevent or dissolve aggregates, although the effect was less potent.
The team then studied neurons derived from Human induced pluripotent stem cells (hiPSCs) from patients with Parkinson's disease (PARK2 mutation) and ALS (TDP-43 mutation). These neurons exhibited reduced cytosolic fluidity, lower ATP levels, and greater protein aggregation than healthy controls.
This supports the idea that ATP deficiency and mitochondrial dysfunction contribute to the condensation of pathogenic proteins in human neurodegenerative diseases.
Implications for Drug Development
This research redefines our approach to therapeutic targets in neurodegenerative diseases. Instead of seeking to eliminate aggregates after their formation, we could:
Target mitochondrial function to preserve ATP production at synapses.
Use small molecules that mimic the hydrotropic effects of ATP to maintain cytoplasmic fluidity.
Develop drugs that prevent the formation of LPS (lipoproteinases) of key disease proteins by improving their solubility.
ATP itself is not a drug molecule in the traditional sense, but these results open new avenues for small molecules capable of acting like ATP to maintain protein solubility or prevent aggregate formation at an early stage.
Conclusion
Neurodegenerative diseases are often viewed from a genetic or protein perspective, but this study provides a biophysical perspective: the physical state of the cytosol itself is crucial. If cells cannot maintain a fluid and soluble environment, primarily due to energy deficiency, aggregation may become inevitable.
This is not just about treating symptoms or even eliminating aggregates afterward. It is about preserving the cellular environment so that neurons can withstand stress and maintain their function. As the field continues to explore how biophysical properties such as viscosity, solubility, and phase separation interact with disease, the role of ATP may prove central, not only as a fuel, but also as a key regulator of neuronal health.
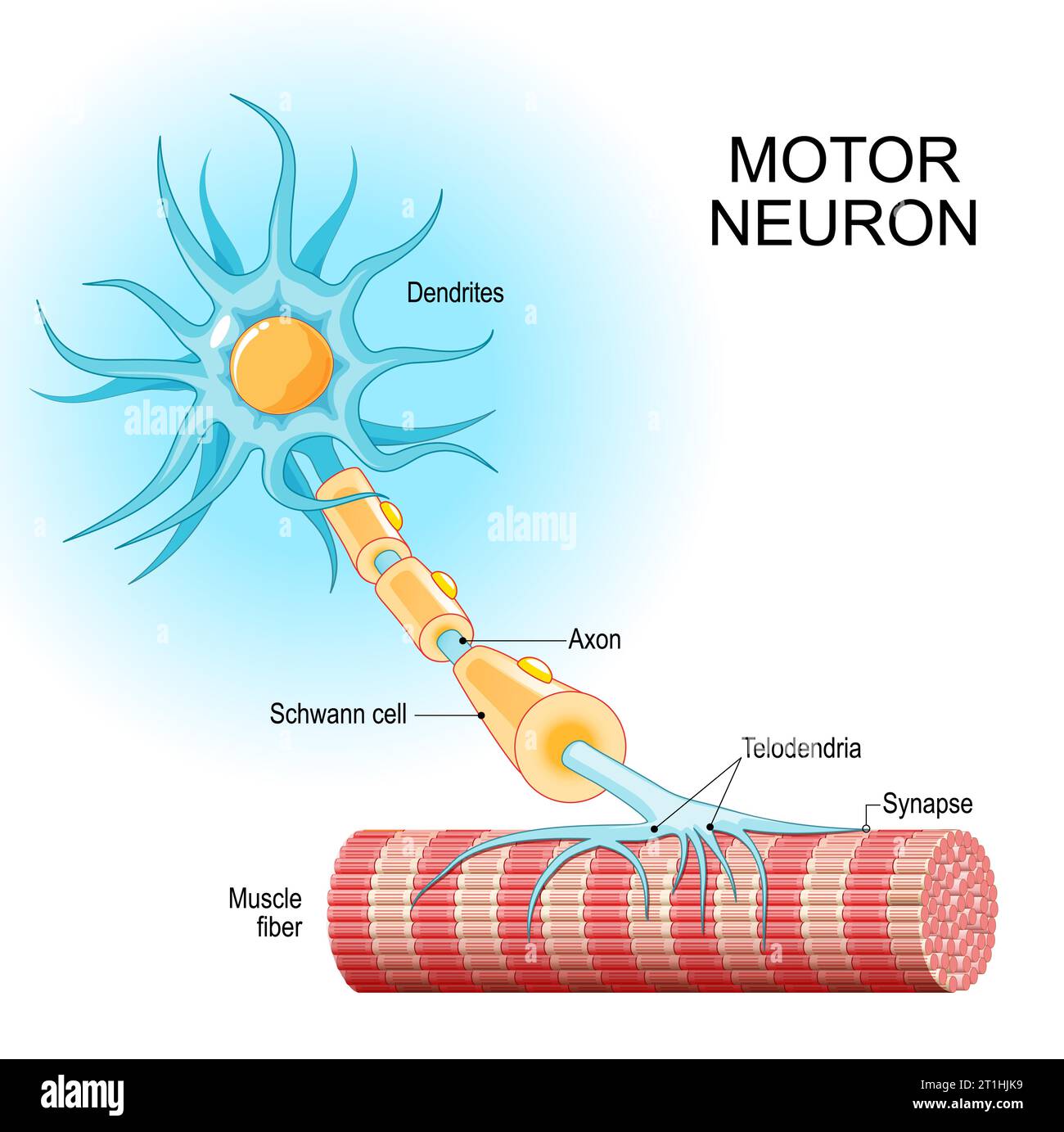
 At this stage, neuroblasts express key transcription factors like ISL1 and LHX3, which establish the fundamental identity of the motor neuron. The neuroblast begins to resemble more to a motor neuron: They extend a long axon out of the spinal cord towards their target muscle. The cell also starts to acquire its specific electrical properties. Then the neuron reaches its target muscle, forms a neuromuscular junction, and becomes a fully functional, electrically active cell. At this point, the early master regulators like ISL1 and LHX3 are largely downregulated, and the neuron enters its final, mature state.
At this stage, neuroblasts express key transcription factors like ISL1 and LHX3, which establish the fundamental identity of the motor neuron. The neuroblast begins to resemble more to a motor neuron: They extend a long axon out of the spinal cord towards their target muscle. The cell also starts to acquire its specific electrical properties. Then the neuron reaches its target muscle, forms a neuromuscular junction, and becomes a fully functional, electrically active cell. At this point, the early master regulators like ISL1 and LHX3 are largely downregulated, and the neuron enters its final, mature state.
 The authors designed a genetic therapy with an AAV virus vector to make mature neurons express two proteins that are only expressed in the immature state.
The AAVs were specifically engineered to target motor neurons. In the study conducted on mice, the administration mode of the AAV viral vector was able to specifically infect the spinal motor neurons.
Once inside the mature motor neurons, the AAV released the therapeutic genes. This caused the neurons to begin expressing ISL1 and LHX3 again
By re-expressing ISL1 and LHX3, the researchers essentially re-activate that original "immature" genetic program. This causes the mature neuron to revert to a state that is genetically and functionally similar to its younger self, with renewed resilience and stress-coping abilities.
They believe that turning on the immature genetic program essentially re-awakens the neuron's dormant ability to regrow and repair itself. While mature neurons in the central nervous system have very limited regenerative capacity, the authors are suggesting that ISL1 and LHX3 could be flipping a switch that bypasses this limitation.
The authors designed a genetic therapy with an AAV virus vector to make mature neurons express two proteins that are only expressed in the immature state.
The AAVs were specifically engineered to target motor neurons. In the study conducted on mice, the administration mode of the AAV viral vector was able to specifically infect the spinal motor neurons.
Once inside the mature motor neurons, the AAV released the therapeutic genes. This caused the neurons to begin expressing ISL1 and LHX3 again
By re-expressing ISL1 and LHX3, the researchers essentially re-activate that original "immature" genetic program. This causes the mature neuron to revert to a state that is genetically and functionally similar to its younger self, with renewed resilience and stress-coping abilities.
They believe that turning on the immature genetic program essentially re-awakens the neuron's dormant ability to regrow and repair itself. While mature neurons in the central nervous system have very limited regenerative capacity, the authors are suggesting that ISL1 and LHX3 could be flipping a switch that bypasses this limitation. De façon similaire, la force des souris modifiés génétiquement est nettement plus basse au début du traitement qu’à la fin, c’est l’inverse de ce qu’on pourrait attendre d’une souris qui serait de plus en plus affaiblie.
Et pourquoi ce groupe de souris aurait-il une force plus faible au début de l’expérience ? S’il n’y a pas de sélection à priori, les différents groupes de souris (traités et non traités) devraient avoir la même force.
De façon similaire, la force des souris modifiés génétiquement est nettement plus basse au début du traitement qu’à la fin, c’est l’inverse de ce qu’on pourrait attendre d’une souris qui serait de plus en plus affaiblie.
Et pourquoi ce groupe de souris aurait-il une force plus faible au début de l’expérience ? S’il n’y a pas de sélection à priori, les différents groupes de souris (traités et non traités) devraient avoir la même force. What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented.
What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented. Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA.
Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA. Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire.
Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire. The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.
The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.