A year ago, a new study drew attention because it was a "hopeful anomaly." It challenged the fatalistic narrative of ALS and provided a clear, new direction for this research field. The ALS therapeutic landscape has seen many failures. Most drugs only slow the disease slightly. For those living with ALS, this study was a concrete reason for hope.
The idea that the secrets to curing the disease might be found in rare individuals who mysteriously recover is a compelling story. It suggests that the biological processes of ALS aren't always one-way and that recovery, though rare, might be possible.
Now, some readers have published a response to this study.
It's behind a paywall, so I haven’t read it, but I can guess what it says. Additionally, a recent publication states: "variants in IGFBP7 were linked to rare "ALS reversals," but the existence of such cases remains controversial." https://pmc.ncbi.nlm.nih.gov/articles/PMC12419016/
In the study published last year, researchers told that they gathered 22 documented reversal cases and validated them across the Target ALS database. This was a pilot case-control study at Duke ALS Clinic in Durham, North Carolina.
The investigators collected demographics, disease details, pedigree info, and saliva samples from ALS reversals. Whole-genome DNA was extracted and sequenced from these saliva samples. The genomes of ALS reversals were then compared to previous whole-genome sequences from a biorepository of de-identified samples of more typical ALS patients. https://clinicaltrials.gov/study/NCT03464903
The researcher has confirmed 34 of these "reversal" cases so far by reviewing medical records. These patients differ in demographics and disease features compared to typical ALS patients. One possible explanation is that these individuals are genetically different, granting them a form of disease "resistance".

However, it appears that cases of ALS reversal are primarily documented in this specialized clinic. This does not mean that such cases do not exist elsewhere, but the diagnosis of ALS and, therefore, of ALS reversal is complicated. For example, it differs between countries in Europe, the United States, and Asia. More importantly, diagnoses vary considerably among doctors.
The problem is that we don’t fully understand what ALS is. Most agree it’s a phenotype that can result from many different causes—some genetic, others from exposure to neurotoxins, physical injury, or other factors.
An example of these variations in practice: the clinical study manager accepted people with primary muscular atrophy (PMA) into his study but PMA is not ALS.
Another issue is how to define “reversal.” Here, reversal was defined as an improvement of at least 4 points on the ALS Functional Rating Scale, maintained for at least 6 months. The ALSFRS-R scale is known to be flawed; it can show improvement simply because of the use of new assistive devices. A 2016 paper co-authored by this researcher stated that most of these “plateaus” and “reversals” are temporary: "ALS plateaus and small reversals are common, especially over brief intervals." https://pubmed.ncbi.nlm.nih.gov/26658909/
The new publication also states: "It is not yet clear if extremely rare “ALS reversals” suffer from typical ALS, or rather from another, yet undescribed disease mimicking ALS diagnostic criteria." https://pmc.ncbi.nlm.nih.gov/articles/PMC12419016/
The last year's study didn't just describe the phenomenon; it identified a specific gene, IGFBP7. It linked reversals to a noncoding variant near IGFBP7, which influences IGF-1 receptor activity. Since IGF-1 has long been suspected of having neuroprotective effects (it has been tested in past ALS clinical trials), this genetic link feels biologically plausible. Yet, more than one hundred genes are associated with ALS, especially SOD1, FUS, and C9orf72 (~9% of cases). These genes aren’t directly related to IGF, making it hard to think that patients with mutations in those genes could still experience reversals.
The authors did openly acknowledge the study’s limitations, which likely sparked discussion:
- The "Reversal" group included only 22 participants. This is a major limitation, and the results must be confirmed with larger groups.
- The study shows a strong genetic link, but it doesn’t prove that the IGFBP7 variant causes the reversals. It seems Professor Bedlack is now exploring this path: https://pubmed.ncbi.nlm.nih.gov/40944442/

 The authors found that key immune signaling proteins, specifically those containing Death Fold Domains (DFDs) (like ASC, FADD, BCL10, MAVS, TRADD), exist in a unique physical state inside our cells called metastable supersaturation. These full-length adaptors retain nucleation barriers and are able to exist supersaturated in cells. In contrast, many receptors and effectors do not. This localizes the “spring-loaded” behaviour to central adaptors that link receptor sensing to downstream cell-fate decisions.
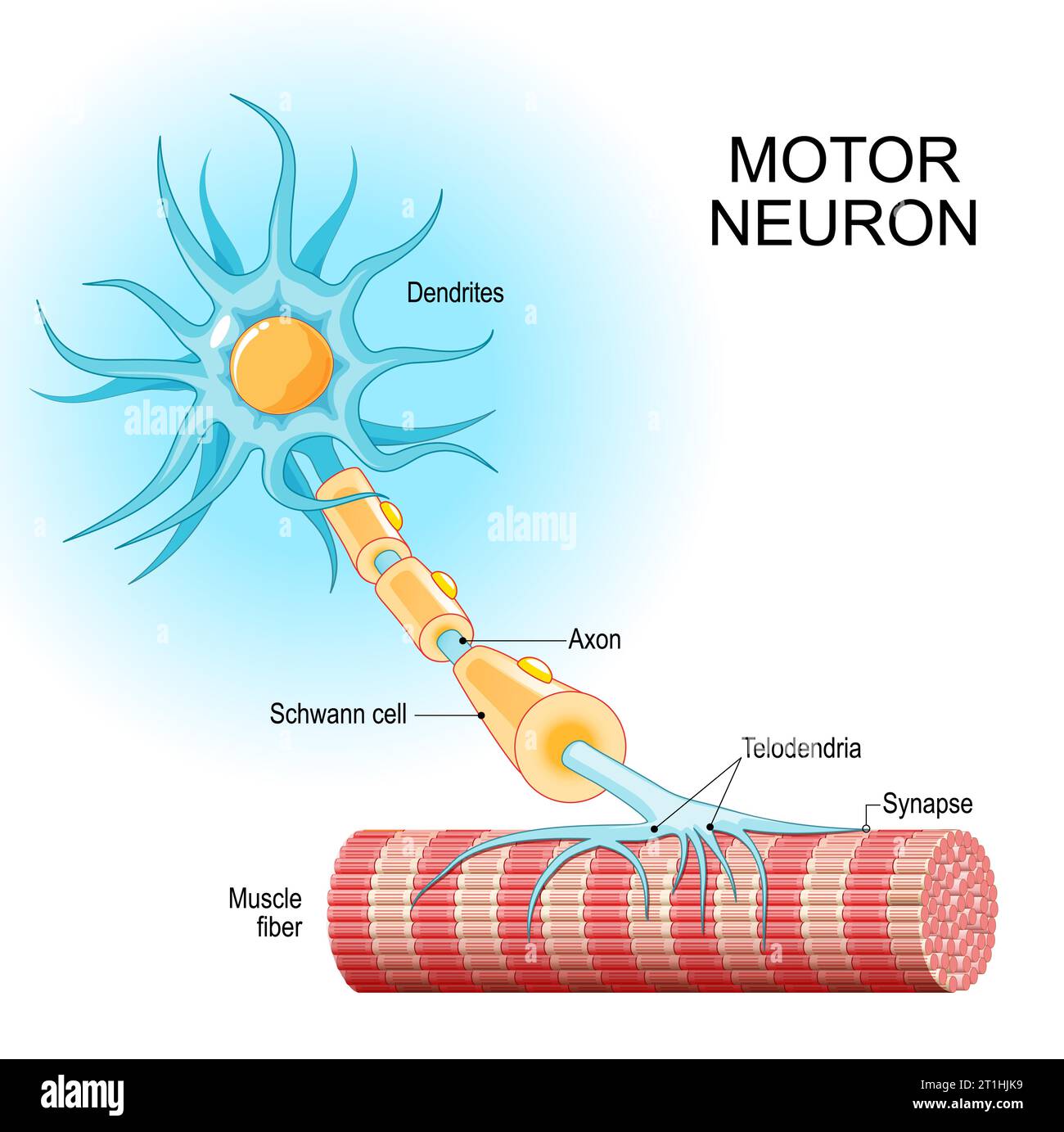
The authors found that key immune signaling proteins, specifically those containing Death Fold Domains (DFDs) (like ASC, FADD, BCL10, MAVS, TRADD), exist in a unique physical state inside our cells called metastable supersaturation. These full-length adaptors retain nucleation barriers and are able to exist supersaturated in cells. In contrast, many receptors and effectors do not. This localizes the “spring-loaded” behaviour to central adaptors that link receptor sensing to downstream cell-fate decisions. At this stage, neuroblasts express key transcription factors like ISL1 and LHX3, which establish the fundamental identity of the motor neuron. The neuroblast begins to resemble more to a motor neuron: They extend a long axon out of the spinal cord towards their target muscle. The cell also starts to acquire its specific electrical properties. Then the neuron reaches its target muscle, forms a neuromuscular junction, and becomes a fully functional, electrically active cell. At this point, the early master regulators like ISL1 and LHX3 are largely downregulated, and the neuron enters its final, mature state.
At this stage, neuroblasts express key transcription factors like ISL1 and LHX3, which establish the fundamental identity of the motor neuron. The neuroblast begins to resemble more to a motor neuron: They extend a long axon out of the spinal cord towards their target muscle. The cell also starts to acquire its specific electrical properties. Then the neuron reaches its target muscle, forms a neuromuscular junction, and becomes a fully functional, electrically active cell. At this point, the early master regulators like ISL1 and LHX3 are largely downregulated, and the neuron enters its final, mature state.
 The authors designed a genetic therapy with an AAV virus vector to make mature neurons express two proteins that are only expressed in the immature state.
The AAVs were specifically engineered to target motor neurons. In the study conducted on mice, the administration mode of the AAV viral vector was able to specifically infect the spinal motor neurons.
Once inside the mature motor neurons, the AAV released the therapeutic genes. This caused the neurons to begin expressing ISL1 and LHX3 again
By re-expressing ISL1 and LHX3, the researchers essentially re-activate that original "immature" genetic program. This causes the mature neuron to revert to a state that is genetically and functionally similar to its younger self, with renewed resilience and stress-coping abilities.
They believe that turning on the immature genetic program essentially re-awakens the neuron's dormant ability to regrow and repair itself. While mature neurons in the central nervous system have very limited regenerative capacity, the authors are suggesting that ISL1 and LHX3 could be flipping a switch that bypasses this limitation.
The authors designed a genetic therapy with an AAV virus vector to make mature neurons express two proteins that are only expressed in the immature state.
The AAVs were specifically engineered to target motor neurons. In the study conducted on mice, the administration mode of the AAV viral vector was able to specifically infect the spinal motor neurons.
Once inside the mature motor neurons, the AAV released the therapeutic genes. This caused the neurons to begin expressing ISL1 and LHX3 again
By re-expressing ISL1 and LHX3, the researchers essentially re-activate that original "immature" genetic program. This causes the mature neuron to revert to a state that is genetically and functionally similar to its younger self, with renewed resilience and stress-coping abilities.
They believe that turning on the immature genetic program essentially re-awakens the neuron's dormant ability to regrow and repair itself. While mature neurons in the central nervous system have very limited regenerative capacity, the authors are suggesting that ISL1 and LHX3 could be flipping a switch that bypasses this limitation. De façon similaire, la force des souris modifiés génétiquement est nettement plus basse au début du traitement qu’à la fin, c’est l’inverse de ce qu’on pourrait attendre d’une souris qui serait de plus en plus affaiblie.
Et pourquoi ce groupe de souris aurait-il une force plus faible au début de l’expérience ? S’il n’y a pas de sélection à priori, les différents groupes de souris (traités et non traités) devraient avoir la même force.
De façon similaire, la force des souris modifiés génétiquement est nettement plus basse au début du traitement qu’à la fin, c’est l’inverse de ce qu’on pourrait attendre d’une souris qui serait de plus en plus affaiblie.
Et pourquoi ce groupe de souris aurait-il une force plus faible au début de l’expérience ? S’il n’y a pas de sélection à priori, les différents groupes de souris (traités et non traités) devraient avoir la même force. What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented.
What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented. Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA.
Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA. Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire.
Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire.