L'accumulation de fer associée au déséquilibre du système d'homéostasie cérébral du fer est une caractéristique pathologique de diverses maladies neurodégénératives. La résonance magnétique a fourni un outil utile pour identifier les maladies neurodégénératives sous-jacente à une concentration anormale du fer dans l'organisme.

FairPark II est un important projet de recherche financé par l'UE qui a étudié principalement les effets d'une thérapie par chélation du fer sur la progression du handicap dans la maladie de Parkinson. Le projet s'est déroulé de 2015 à 2020 et a réuni 15 partenaires dans un essai clinique multicentrique de la thérapie chez les patients atteints de la maladie de Parkinson. FAIR-ALS-II est un projet français similaire à FairPark II, mais pour la sclérose latérale amyotrophique.
Dans les trois principaux maladies neurodégénératives, la dégénérescence se produit dans les régions du système nerveux central (SNC) associées à la mémoire (maladie d'Alzheimer), à l'automaticité (maladie de Parkinson) et à la fonction motrice (sclérose latérale amyotrophique, sclérose latérale amyotrophique), qui nécessitent toutes une demande en oxygène pour exploiter les besoins importants en énergie de ces neurones.
Dans la maladie de Parkinson, une dégénérescence progressive de la substantia nigra pars compacta (SNc) est associée à l'apparition de foyers sidérotiques [21], en grande partie causée par une augmentation instable des niveaux de fer résultant d'un déséquilibre entre l'importation, le stockage et l'exportation de fer cellulaire. Au niveau moléculaire, l'α-synucléine régule le transport de la dopamine et du fer avec des mutations associées à la maladie de Parkinson dans cette protéine, provoquant une perturbation fonctionnelle de ces processus.
De même, dans la sclérose latérale amyotrophique, une accumulation précoce de fer est présente dans les neurones de la voie motrice cortico-spinale avant l'apparition de la maladie et avant l'accumulation secondaire de fer dans la microglie. Un taux élevé de ferritine dans le sérum est un indicateur de mauvais pronostic pour les malades de la sclérose latérale amyotrophique et l'application de séquences sensibles au fer en imagerie par résonance magnétique est devenue un outil utile pour identifier cette maladie.
Les voies moléculaires qui découlent d'un tel déséquilibre du système d'homéostasie restent encore à élucider, mais des percées importantes ont été réalisées ces dernières années. Loin d'être une simple cause ou conséquence, il a été récemment découvert que ces altérations peuvent déclencher une sensibilité à une voie de mort cellulaire dépendante du fer qui est appelée ferroptose. À son tour, cela a entrainé un intérêt pour certains modulateurs clés de cette voie de mort cellulaire qui pourraient être des cibles thérapeutiques pour les maladies neurodégénératives.
Il est intéressant de remarquer que l'accumulation de fer et la ferroptose sont très sensibles à la chélation du fer. Cependant, bien que les chélateurs qui récupèrent le fer intracellulaire, protègent contre les dommages neuronaux oxydatifs dans les modèles mammifères et se sont révélés efficaces pour traiter la sidérose systémique, ces composés ne sont pas appropriés en raison du risque élevé de développer une déplétion en fer et une anémie. Au lieu de cela, une chélation modérée du fer offre une nouvelle stratégie thérapeutique pour la neuroprotection. Comme le démontre le défériprone, le fer peut être récupéré des complexes de fer instable dans le cerveau et transféré (de façon conservatrice) vers des accepteurs d'affinité plus élevée dans les cellules ou la transferrine extracellulaire. Des essais de preuve de concept précliniques et cliniques prometteurs ont conduit à plusieurs essais cliniques de grande envergure.
[21] "sidérotique" signifie: Lié au fer ou à l'acier, comme dans la sidérose (fibrose causée par des dépôts de fer)

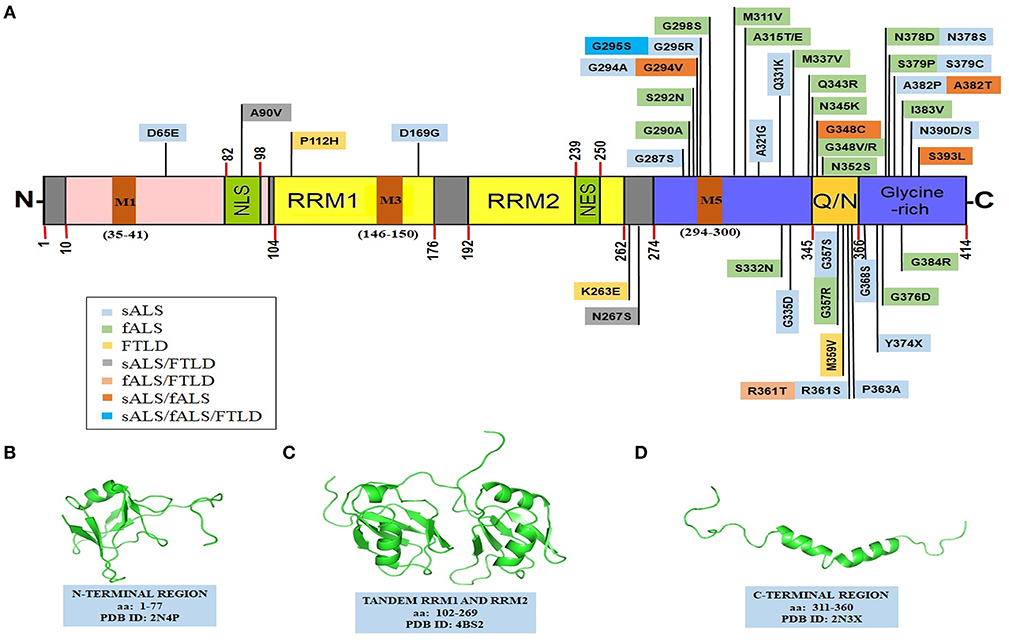 Les auteurs ont montré qu'en présence d’une acidification, même très faible, le TDP-43tRRM se replie complètement et s'oligomérise pour former une «forme β» riche en feuillets β. La forme β a une structure ordonnée et stable qui ressemble aux fibrilles amyloïdes que l'on trouve dans la maladie d'Alzheimer!
Les auteurs ont montré qu'en présence d’une acidification, même très faible, le TDP-43tRRM se replie complètement et s'oligomérise pour former une «forme β» riche en feuillets β. La forme β a une structure ordonnée et stable qui ressemble aux fibrilles amyloïdes que l'on trouve dans la maladie d'Alzheimer!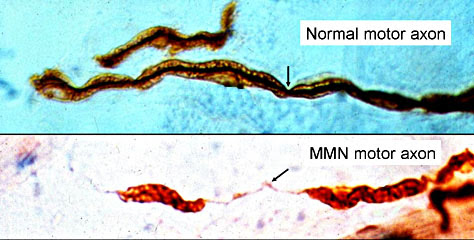 La nature exacte des problèmes apparaît cependant être variable dans ces différentes maladies; par exemple les concentrations de ganglioside
sont réduites dans la maladie de Parkinson et la maladie de Huntington, mais augmentées dans la maladie d’Alzheimer et y a des altérations dans les deux directions pour la SLA.
La nature exacte des problèmes apparaît cependant être variable dans ces différentes maladies; par exemple les concentrations de ganglioside
sont réduites dans la maladie de Parkinson et la maladie de Huntington, mais augmentées dans la maladie d’Alzheimer et y a des altérations dans les deux directions pour la SLA.
 Par BrainsRusDC - Travail personnel, CC BY 4.0, https://commons.wikimedia.org/w/index.php?curid=64271015
Par BrainsRusDC - Travail personnel, CC BY 4.0, https://commons.wikimedia.org/w/index.php?curid=64271015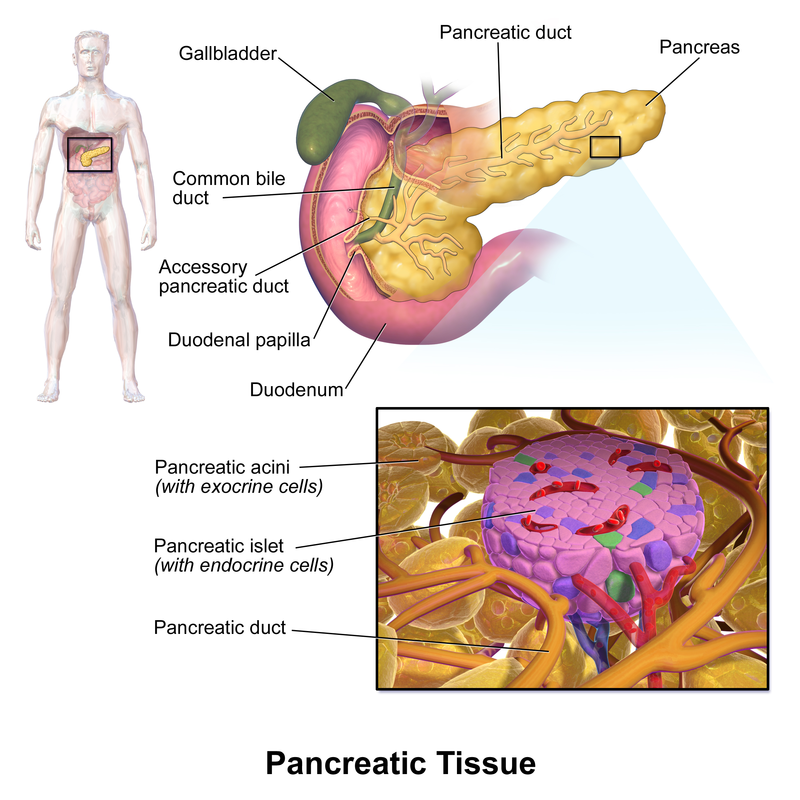 Source: Blausen.com staff (2014). "Medical gallery
Source: Blausen.com staff (2014). "Medical gallery